Preparado por: Dr. José Antonio Martínez Oyanedel
Departamento de Biología Molecular, Facultad de Ciencias Biológicas
Proyecto Docencia 97-003
CAPITULO 3: ESTRUCTURA SECUNDARIA Y CONFORMACIÓN DEL ESQUELETO
ANGULOS DE TORSIÓN
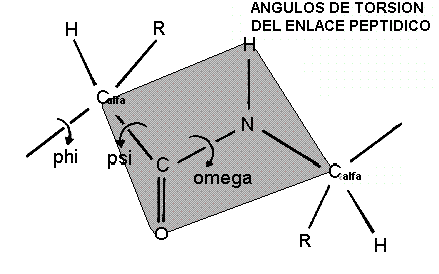
La figura de abajo muestra los tres ángulos de torsión principales del enlace peptídico o de un polipéptido: phi (
F), psi (Y), y omega (W).
La planaridad del enlace peptídico es de 180º en casi todos los enlaces peptídicos de la cadena principal. Solo en raros casos es de 10º para un enlace peptídico cis el cual implica un residuo de prolina.
GRÁFICO DE RAMACHANDRAN
En la cadena polipeptídica los enlaces N-C
a y Ca-C tienen libre rotación. Estas rotaciones están representadas por los ángulos de torsión phi (F) y psi (Y), respectivamente.GN Ramachandran, científico hindú, uso modelos de pequeños polipéptidos para variar sistemáticamente estos ángulos con el objeto de encontrar conformaciones estables. Para cada una de las conformaciones, la estructura fue examinada para detectar los contactos cercanos entre los átomos. Los átomos fueron considerados como esferas rígidas con dimensiones correspondientes a su radio de van der Waals. Por lo tanto, los ángulos que causaban una colisión de las esferas correspondían a ángulos prohibidos y correspondían a conformaciones no permitidas de la cadena polipeptídica.
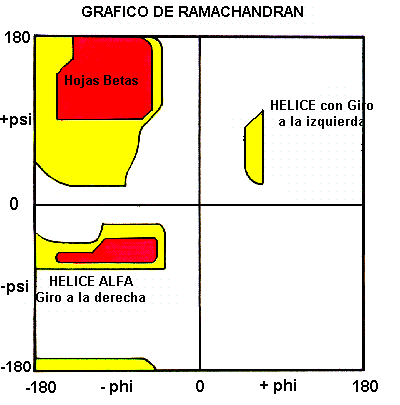
En el diagrama anterior las zonas blancas corresponden a conformaciones donde los átomos del polipeptido están más cercanos que sus radios de van der Waals. Estas regiones son estericamente no permitidas para todos los aminoácidos, excepto para Glicina, el cual es el único aminoácido que no presenta cadena lateral. Las regiones rojas corresponden a conformaciones donde no hay impedimentos estericos, es decir estas son zonas permitidas llamadas conformaciones
a-hélices y Hojas-b. Las zonas amarillas muestran las regiones permitidas sí en los cálculos se usan radios van der Waals ligeramente más pequeños, es decir se permite que los átomos puedan estar un poco mas cercanos. Esto origina una nueva región que corresponde a una hélice con giro hacia la izquierda. Los L-aminoácidos no pueden formar una hélice con giro a la izquierda pero ocasionalmente residuos individuales pueden adoptar esta conformación. Estos residuos generalmente son glicina pero también pueden ser asparragina y acido aspártico en los que la cadena lateral forma un puente hidrógeno con la cadena principal y así estabiliza esta conformación desfavorable. La hélice 310 se presenta en el extremo superior-derecho de la región helicoidal y esta en el extremo de la región permitida lo que indica su baja estabilidadLas regiones no permitidas generalmente involucran impedimentos estericos entre el carbono metilénico de la cadena lateral y los átomos de la cadena principal. La Glicina no tiene cadena lateral y por lo tanto puede adoptar ángulos phi y psi en los cuatro cuadrantes del gráfico de Ramachandran. Por esto ellas aparecen en regiones de vueltas de la proteína donde cualquier otro residuo estaría estericamente impedido.
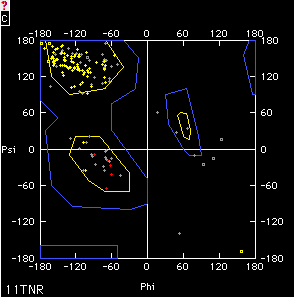
Abajo se presenta un gráfico de Ramachandran de una proteína que contiene casi exclusivamente hojas betas (puntos amarillos) y solo una hélice (puntos rojos). Note que solo algunos residuos están fuera de las regiones permitidas; siendo la mayoría de ellos Glicinas ( marcados como un cuadrado en vez de una cruz).
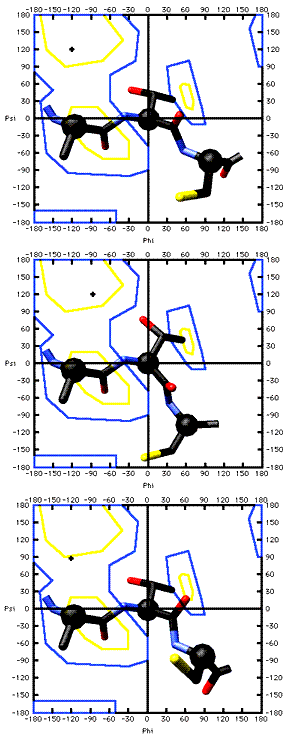
Observe el efecto de cambios menores en los ángulos Phi y Psi: primero para phi -120, psi 120; luego un cambio en phi, phi -90, psi 120; ahora un cambio en psi, phi -120, psi 90.
ESTRUCTURA SECUNDARIA
Las diferentes zonas de la cadena polipeptídica pueden adoptar ordenamientos regulares de sus ángulos phi y psi, lo que origina las llamdas estructras secundarias regulares. Se describen tres tipos de estas estructuras: Las hélices, las estructuras extendidas y las vueltas.
LA
a-HÉLICE.Desarrollo de un modelo de estructura de una
a-hélice.Pauling y Corey tomaron cadenas polipeptídicas modelos y los plegaron de forma de obtener plegamientos regulares de la cadena de modo que pudieran explicar los datos de difracción de la
a-queratina . El plegamiento más simple y elegante fue una conformación en espiral a la derecha conocida como a-hélice.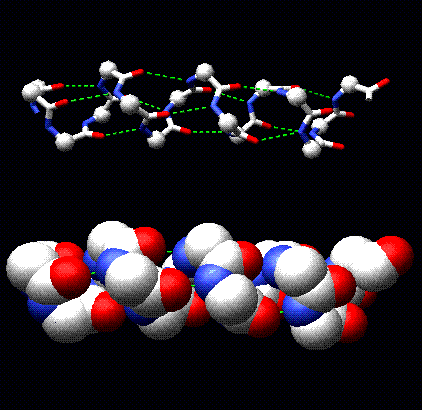
Propiedades de la
a-hélice.La estructura se repite cada 5.4 Å a lo largo del eje de la hélice, es decir, diremos que la hélice tiene un paso de 5.4 Å. Las
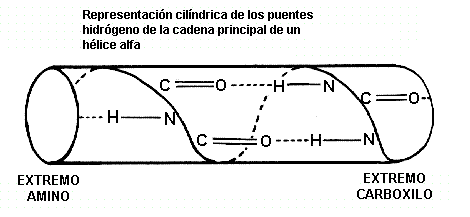
a-hélices tienen 3.6 residuos de aminoácidos por vuelta, es decir una hélice con 36 residuos podría formar 10 vueltas. La separación de los residuos a lo largo del eje de la hélice es 5.4/3.6 o 1,5; en otras palabras la hélice tiene un avance de1.5 Å.Cada grupo C=O y N-H de la cadena principal establece un puente hidrógeno con un enlace peptídico 4 residuos por adelante. (i.e. Oi a N i+4). Esto da un ordenamiento estable.
Los planos del enlace peptídico están semi paralelos con el eje de la hélice y los dipolos dentro de la hélice están alineados, es decir todos los grupos C=O apuntan en la misma dirección y todos los grupos N-H apuntan en la otra dirección. Las cadenas laterales apuntan afuera de la hélice y generalmente se orientan hacia el extremo N-terminal de la hélice.
Todos los aminoácidos tienen phi y psi con valores negativos, valores típicos son -60º y -50º, respectivamente.
Distorsiones en la
a-hélice.La mayoría de las
a-hélices en proteínas globulares están curvadas o distorsionadas si se comparan con el modelo estándar de Pauling-Corey. Estas distorsiones se originan por varios efectos, entre los cuales se incluyen: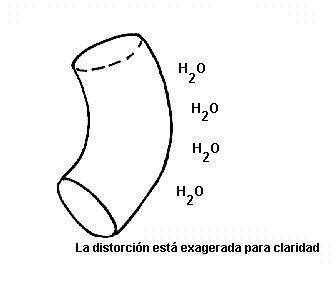
HÉLICES 310.
Estrictamente estás forman una clase de hélices distintas pero son cortas y frecuentemente aparecen en la región terminal de las
a-hélices. El nombre El nombre 310 se origina porque hay tres residuos por vuelta y diez átomos forman el anillo que se forma con cada puente hidrógeno (nótese que también se incluye el Hidrógeno formando parte del anillo). Hay puente hidrógeno entre los átomos de la cadena principal de residuos separados por tres unidades a lo largo de la cadena ( es decir, Oi a N i+3). En esta nomenclatura de Pauling-Corey la a-hélice es una hélice 3.613-.Los dipolos de la hélice 310 no están alineados como en la a-hélice, es decir es una estructura menos estable y el empaquetamiento de las cadenas laterales es menos favorable.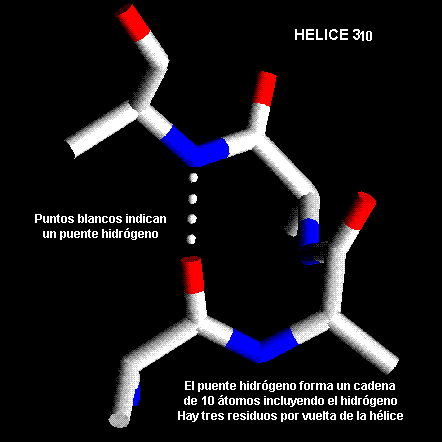
Existe otro tipo de hélice la llamada hélice
p, que corresponde la hélice 416.
LA ESTRUCTURA
bPauling y Corey encontraron un modelo para las estructuras de proteínas fibrilares conocidas como
b-queratinas. En esta conformación el polipeptido no forma una estructura al azar, por el contrario forma un zigzag en una conformación más extendida que la a-hélice. Los residuos de aminoácidos en la conformación b presentan ángulos F negativos y ángulos Y positivos. Los valores típicos son F = -140º y Y = 130º. Por el contrario los residuos en conformación a-hélice tienen ambos F y Y negativos. Una sección del polipeptido con residuos en conformación b, es llamado como una hebra-b y estas hebras pueden asociarse por medio de puentes de hidrógeno para formar una hoja-b.En una hoja-
b dos o más cadenas polipeptidicas corren a lo largo una de la otra y están unidas de forma regular por puentes de hidrógeno entre los grupos C=O y N-H de la cadena principal. Por lo tanto los puentes de hidrógeno en una hoja-b son entre diferentes segmentos de la cadena polipeptidica. Esto contrasta con la a-hélice donde los puentes de hidrógeno involucran el mismo elemento de estructura secundaria, es decir una misma zona de la cadena polipeptídica. En la hoja-b los grupos R, o cadenas laterales de los residuos vecinos apuntan en direcciones opuestas.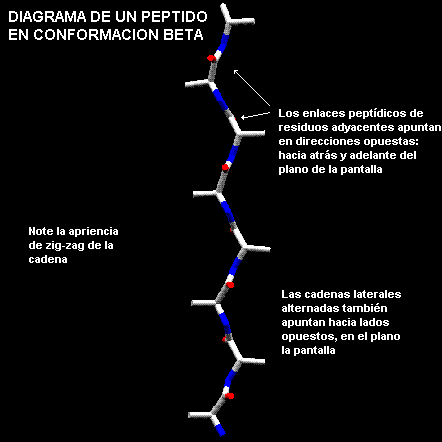
Imaginemos dos hebras paralelas a esta, una sobre el plano de la pantalla y otra atrás, es posible visualizar como se origina la apariencia plegada de la hoja. Note que los grupos del enlace peptídico de residuos adyacentes apuntan en direcciones opuestas mientras que en las hélices los enlace peptídicos apuntan en el mismo sentido.
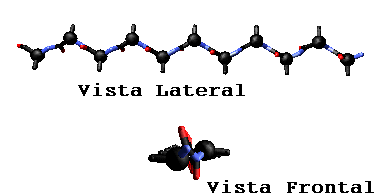
La distancia axial entre los residuos adyacentes es 3.5 Å. Hay dos residuos por unidad de repetición los cual le da a la hebra-
b una periodicidad de 7 Å. Esto comparado con la a-hélice donde la distancia axial entre residuos adyacentes es solamente 1.5 Å nos muestra claramente que los polipeptidos en conformación b son bastante más extendidos que los polipeptidos en conformación helicoidal.Hojas-
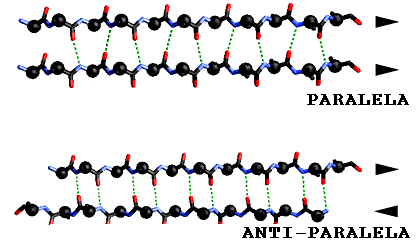
b Paralelas, anti-paralelas y mixtas.En las hojas-
b las hebras corren en una dirección mientras que en las hebras anti-paralelas estas corren en direcciones opuestas. En las hojas mixtas algunas hebras están paralelas y otras anti-paralelas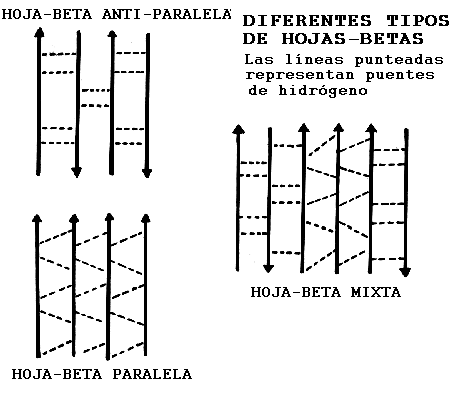
Abajo se presenta un diagrama de una hoja-
b de ocho hebras antiparalelas, que esta formada por dos zonas de la proteína. Se enfatiza el alto orden y la regularidad de los puentes de hidrógeno entre NH y CO de las cadenas principales de las hebras constituyentes.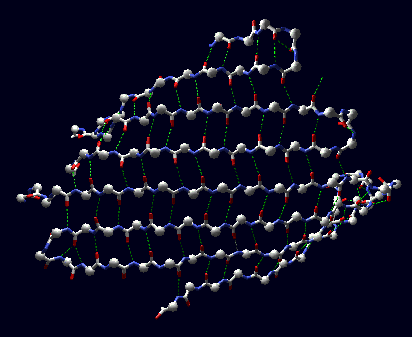
En los modelos clásicos de Pauling-Corey las hojas paralelas presentan los puentes hidrógeno más distorsionados y consecuentemente más débiles entre las hebras:
Las hebras-
b son muy comunes en las proteínas globulares y la mayoría contiene menos de 6 hebras. El ancho de una hoja-b de seis hebras es de aproximadamente 25 Å. No se observa preferencia por hebras paralelas o antiparalelas, pero las hebras paralelas con menos de cuatro hebras son raras, quizás reflejando su baja estabilidad. Las hojas tienden a ser paralelas o antiparalelas, pero también se presentan hojas mixtas.El modelo de Pauling-Corey de las hojas-
b es plano. Sin embargo, la mayoría de las hojas-b encontradas en las estructuras cristalinas proteínas globulares son torcidas. Esta torsión es hacia la izquierda como se muestra en este esquema . La torsión total de la hoja resulta de la rotación relativa de cada residuo de la hebra en 30 grados en sentido horario (hacia la derecha).
Las hojas paralelas son menos torcidas que la hojas antiparalelas y siempre están internas. Por el contrario, las hojas antiparalelas pueden soportar mayores distorsiones (torsiones y nudos-
b) y son más expuestas al solvente. Esto implica que las hojas antiparalelas son más estables que las paralelas lo cual es consistente tanto con la geometría de los puentes de hidrogeno como con el hecho que hebras paralelas pequeñas son escasas.
VUELTAS REVERSAS
Una vuelta reversa es la región de la cadena polipeptidica que tiene un puente de hidrógeno desde el oxígeno del grupo carbonilo de la cadena principal hasta el grupo NH del residuo que se encuentra 3 posiciones más adelante en la cadena ( es decir Oi a Ni+3). Las regiones helicoidales están excluidas de esta definición y las vueltas entre las hebras-
b forman una clase especial de vueltas conocidas como horquillas-b (ver capítulo siguiente). Las vueltas reversas son muy abundantes en las proteínas globulares y generalmente aparecen en la superficie. Se ha sugerido las regiones de vueltas actúan como centros de nucleación durante el plegamiento de las proteínas.Las vueltas reversas están divididas en clases de acuerdo al valor de los ángulos
F y Y de los residuos en las posiciones i+1 e i+2. Los tipos I y II que se muestran en la figura de más abajo son las vueltas reversas más comunes la diferencia esencial entre ellas es la orientación del enlace peptídico entre los residuos i+1 e i+2.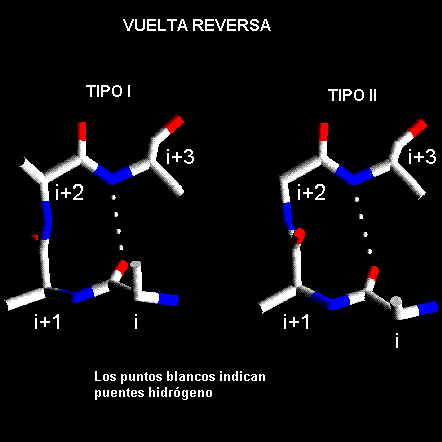
Los ángulos de torsión para los residuos i+1 e i+2 en los dos tipos de vueltas están en distintas regiones del gráfico de Ramachandran.
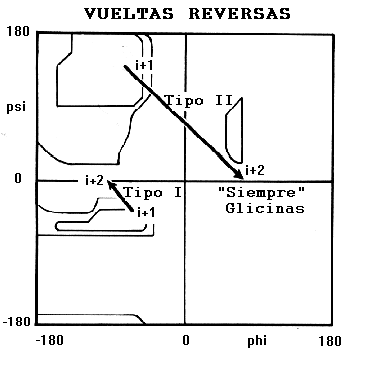
Note que el residuo i+2 de la vuelta reversa del tipo II cae en una región del gráfico de Ramachandran que solo puede ser ocupado por residuos de glicina. A partir del diagrama de este tipo de vuelta se puede ver que si el residuo i+2 posee cadena lateral, esta podría presentar impedimentos estéricos con el oxígeno carbonílico del residuo precedente. De esto se deduce que el residuos i+2, en la vueltas reversas del tipo II, siempre es glicina.