TÓPICOS EN FARMACOCINÉTICA
Volumen de
Distribución
- El volumen de distribución (Vd) es la razón
entre la cantidad de fármaco en el organismo y la concentración
del fármaco en el plasma o sangre (C).
- Vd = (cantidad de fármaco en el organismo)/C donde
C es la concentración de fármaco en el plasma o
sangre.
- Vd como es calculado, es un volumen
aparente de distribución. Por ejemplo:
- Vd para digoxina
es 440 L/70 kg
- Vd para cloroquina es 13,000 L/70 kg
- Este gran Vd podría ser consistente com
una elevada unión a tejidos, dejando poco
fármaco libre en el plasma o sangre.
- Vd es un volumen aparente de distribución
puesto que es el volumen de líquido corporal que
contiene la cantidad de fármaco a la concentración
encontrada en el plasma, sangre o agua plasmática
y homogéneamente distribuído.
- Muchos fármacos tienen mayor concentración en los
compartimentos extravasculares, por tanto estos
agentes NO están homogéneamente distribuídos
en el organismo.
Volumenes físicos (L./kg peso corporal) para
algunos compartimentos
Agua
| Total
de agua corporal (0.5-0.7 L/kg) o aprox. 35000
a 49000 ml (individuo de 70 kgl) |
Agua
extracelularr (0.2 L./kg) |
Sangre (0.08 L./kg);
Plasma (0.04
L./kg) |
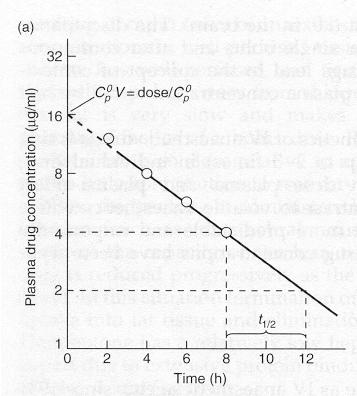
Grafico semilogarítmico
en que se muestra a extrpolación a tiempo=0
la concentarción
plasmática
(Co),
requerida para determinar el volumen de distribución:
Vd = dose/Co. Observe
que de este tipo de gráfico también puede determinarse
el tiempo de vida media de eliminación del fármaco.
Este gráfico supone que el fármaco se distribuye en un único
compartimento.
- Factores que influyen en el volumen de
distribución:
- grado de unión a proteínas plasmáticas
- coeficiente de partición lípido/agua
- El Vd puede se afectado por:
- edad del paciente
- enfermedad del paciente
- composición corporal del paciente
- El Clearance es importante
para asegurar una correcta dosificación, especialmente
en el caso de dosis repetidas.
- El
Clearance de un determinado fármaco es generalmente
constante, en el rango de concentración terapéutica
debido a que :
- Los sistemas de eliminación
de fármacos no están saturados, por lo tanto la
velocidad de eliminación es una función lineal de
la concentración plasmática.
- La eliminación de fármacos , por
lo tanto, sigue, usualmente, una cinética de
primer orden,i.e, una fracción constante del
fármaco es eiliminada por unidad de tiempo.
- Algunos fármacos, porejemplo el etanol,
presentan una cinética de orden cero: una cantidad
constante es eliminada en la unidad de tiempo. {El
Clearance es variable}
- Clearance: la velocidad de eliminación de
un fármaco (por todas las vías) normalizada a la
concentración (C) del fármaco en algún fluído biológico.
- volumen por unidad de tiempo (volumen de fluído i.e.
sangre o plasma que es completamente depurado del fármaco en la unidad
de tiempo)
- puede ser definido como:
- clearance sanguíneo, CLb
- clearance plasmático, CLp
- El Clearance es aditivo: función de la eliminación
por todos los órganos participantes, tales como
el hígado o rińón.
"Otros" en
la ecuación anterior puede incluir los
pulmones y otros sitios de metabolismo de
fármacos tales como la sangre misma.
- Los rińones son los órganos más importantes
para la eliminación del fármaco propiamente
tal y de los metabolitos del fármaco.
- Loscompuestos solubles en agua presentan
una excreción renal más eficiente comparados
con los liposolubles.
- El clearance renal de un fármaco se correlaciona
con el clearance de creatinina exógena o
con la concentración sérica de creatinina.
1.-
Filtración glomerular :
2.-
Secreción tubular (proceso activo)
3.- Reabsorción tubular pasiva:
- Una alta liposolubilidad favorece
la reabsorción de fármacos (los
compuesto liposolubles se ven favorecidos
pra atravesar las membranas celulares
de las células epiteliales renales
y entrar así al fluído pericapilar)
- La velocidad de reabsorción tubular
está influenciada por:
- pH
- velocidad del flujo urinario tubular
renal
- pKa del fármaco o su metabolito
comparado con el pH de la orina.
La velocidad de filtración depende de:
- volumen filtrado en el glomérulo
- concentración
del fármaco unido en el plasma. (el fármaco
unido a proteínas plasmáticas no es
filtrado)
La llegada del fármaco
a los sitios de eliminación puede ser la velocidad
limitante para muchos de ellos:
- en la llamada eliminación "flujo-dependiente"
la mayoría del fármaco en la sangre es
eliminada en un paso de la sangre por el
órgano de eliminación.
- estos fármacos tienen una elevada
razón de extracción
Clearance > 6 ml/min./kg --
incluye:
|
|
|
Cambios en el clearance intrínseco
(i.e. inducción enzimática, enfermedad hepática, : afecta el clearance
con baja razón de extracción)
- Factores sociales:
- El humo de cigarrillo induce algunas
isoformas de las enzimas metabolizantes
de fármacos (CYP1A1, CYP1A2, y posiblemente CYP2E1)
- Consideraciones dietarias:
- El jugo de ciertas frutas (pomelo)
contiene agentes químicos que son potentes
inhibidores de la CYP3A4 localizada
en la mucosa de la pared intestinal..
- El calcio presente en diversos alimentos
o productos pudede quelar fármacos como
las tetraciclinas y fluoroquinonas.
- Edad: Los neonatos presentan una
disminuída exreción renal y metabolismo
hepático debido a la inmadurez de
estos órganos. Por otro lado, los
ancianos, presentan diferencias en la absorción,
metabolismo hepático, clearance renal,
metabolismo hepático y volumen de
distribución.
- Factores genéticos:
- El polimorfismo genético que afecte
CYP2D6,
CYP2C19, CYP2A6, CYP2C9, y N-acetyltransferasa da como resultado
una
significtiva diferencia interindividual en la habilidad de metabolizar
fármacos, simpre que éstos sean sustratos
de agunas de estas isoformas del citocromo
P450.
- Ciertos polimorfsmos genéticos están
asociados con grupos etnicos.Por
ejemplo, entre 5%-10% de los cacucásicos
son pobres metabolizadores de sustratos
de la CYP2D6, la frecuencia en la población
asiática es de
1%-2%. Por otro lado la incidencia de bajos metabolizadores
de fármacos que lo hacen a través
de laf CYP2C19, es de 20% en
poblaciones asiáticas y 4% en caucásicos.
Citocromo P450 :convenciones para
nombrar las isoformas:
La biotransformación de fármacos
compromete, generalmente, dos fases:
Fase I y Fase II.
- Las reacciones de Fase I son comprende
las oxidaciones, reducciones
o hidrólisis que sufre el fármaco. Los
metabolitos resultantes de esta
fase son generalmente más polares,
incrtementandose la posibilidad
de estos de ser excretados por
el rińón. Estos metabolitos
pueden ser posteriormente metabolizados
nuevamente.
- Los
metabolitos de la Fase I son
generalmente sustratos de la
Fase II que cataliza la adición
de otros grupos, por ejemplo,
acetatos, gucuronatos, sulfato
o glicina a los grupos polares
presentes en el intermediario.
Después de las reacciones de
Fase II el metabolito resultantes
e mucho más excretable.
- La
mayoría de las reacciones de
la Fase I son catalizadas por
el sistema citocromo P450
(CYP). Esta superfamilia consiste
en isoenzimas que contienen
hem, que está localizadas en
los hepatocitos, específicamente
en el retículo endoplásmico
liso El principal sitio extrahepático
que contien isoformas del CYP
podrían ser los enterocitos
del intestino delgado.
- El nombre de la familia de genes
es especificada por un número arábigo e.g. CYP3.
- Las familias CYP están subdivididas
en subfamilias designadas por
una letra mayúscula, e.g.
CYP3A .
- El múmero de genes de las enzimas
individuales son especificadas
por un segundo número arábigo, e.g. CYP3A4.
- Las CYP isoformas metabolizan
sustancias endógenas como
prostaglandinas, lípidos, ácidos grasos, y hormonas esteroidales,
también metabolizan o detoxifican
sustancias exógenas, incluyendo
fármacos.
- Las principales isoformas de CYP,
responsables del metabolismo de
fármacos son: CYP3A4, CYP2D6, CYP2C9, CYP2C19,
CYP1A2, CYP2E1 y en ciertos casos CYP2A6 y CYP2D6.
- Si el flujo al órgano no es la limitante
de la eliminación, entonces la relación entre
la velocidad de eliminación del fármaco y la
concentración (C) de éste es:
- la forma de esta ecuación es muy similar
a la de Michaelis-Menten, para cinética enzimática. Aquí,
sin embargo:
- Km es
la concentración del
fármaco al cual la velocidad de eliminación
es un 50% de Vmax.
- Como se espera a partir de la forma hiperbólica
de la curva, a altas concentraciones del
fármaco (comparada a la Km), la influencia
de la concentración del fármaco sobre la
velocidad de eliminación disminuye significativamente,
aproximandose a un comportamiento
de orden cero.:
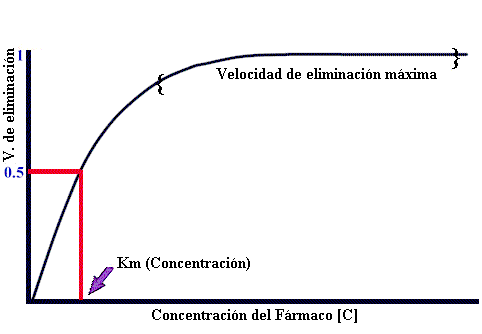
Tiempo de vida media
- El tiempo de vida media: (t1/2) -- es
el tiempo requerido para que una cantidad
de fármaco en el organismo disminuya a la
mitad de su valor durante la eliminación (o durante
una infusión constante).
- Supongamos:
- un único compartimento de tamańo igual al volumen
de distribución (Vd)
- sangre o plasma en equilibrio con el volumen de
distribución
- t1/2 = (0.693 · Vd)/CL
- t1/2 = (0.693)/kel
- kel = km + kex; , kel es
la constante de eliminación e
igual a la suma de las constantes
de velocidad de metabolización, km , y
de
excreción, kex.
- Factores que afectan el t1/2:
- enfermedades-- afectan el volumen de distribución
y/o el clearance
ejemplo 2: vida media
del diazepam aumenta con la edad
- el clearance no cambia
- el volumen of distribución cambia
ejemplo 3:
cambio secundario en el tiempo de vida
media por cambios en la unión a proteínas.
- En pacientes con hepatitis viral el tiempo de
vida media de la Tolbutamida disminuye.
- En la hepatitis viral aguda la unión a proteínas
plasmáticas y tejidos; no se altera
el Vd, pero aumenta el clearance
debido al incremento de fármaco
no-unido (libre)
- Tiempo de vida media de eliminación y anestesia:
- El tiempo de vida media de eliminación es importante
para estima el tiempo de recuperación después
de la administración del fármaco anestésico.
- En el caso de agente administrados por vía endovenosa
hay una inconsistencia entre las vidas
medias después de una única administración
comparada conn la obtenida por una infusín
endovenosa contínua, lo que ha resultado
en la idea de referirlas como "sensibles
al contexto" o vidas medias dependientes..
- La definición de vidas medias "sensibles al
contexto" es definida como como
el tiempo requerido para que la concentración
sanguínea del fármaco caiga al 50% después
de una infusión contínua.
- Este problema que se observa con naestésicos IV,
parece no existir para aquellos anestésicos
volátiles.
- Vida media
- Util para calcular el estado estable. Aproximadamente
4 vidas medias son requeridas para obtener
un 94% de un nuevo estado estable.
- Util para estimar el tiempo requerido para que se
elimine un fármaco desde el organismo
- Util para estimar los intervalos de dosificación
apropiados.
Acumulación de fármacos
- Con la administración de dosis repetidas, el fármaco
se acumula en el organismo hasta que cese su
administración.
- En fórma práctica, la acumulación se observa
cuando el fármaco es administrado repetidamente
a intervalos menores de 4 vidas medias.
- La acumulación es inversamente proporcional a la fracción
de la dosis perdida en cada intervalo.l
Biodisponibilidad
- Definición: fracción del fármaco no modificado,
que alcanza la circulación sistémica después
de su administración.(por cualquiera vía de
administración)
- Ejemplos:
- biodisponibilidad por vía EV=1
- Otras vías de administración = < 1
- Principales factores que reducen la biodisponibilidad
a menos de 100%
- absorción incompleta
- efecto de primer paso ( metabolización por el hígado
antes que el fármaco llegue a la circulación
general)
- Una absorción incompleta después de la administración
oral del fármaco (hecho frecuente)
- Por ejemplo sólo el 70% de la digoxina alcanza la
alcanza la circulación sistémica:
- Factores:
- baja absorción gastrointestinal
- metabolización de la digoxina por la flora
gastrointestinal
- Fármaco muy hidrofílico - no bien absorbido- pue
no puede atravesar el componente lipídico
de la membrana celular.
- Fármacos excesivamente liposolubles: incapaces de
atravesar la capa acuosa cercana a la membrana
lipídica.
- Secuencia de transporte:
- paso a través de la pared intestinal hacia la circulación
portal
- transporte del fármaco por la porta hacia el hígado
- el fármaco puede alcanzar la circulación sistémica
- la biodisponibilidad puede ser afectada
por alguno de estos pasos
- metabolización del fármaco puede ocurrir en la pared
intestinal y/o sangre
- metabolización del fármaco (potencialmente
alto) puede ocurrir en el hígado
- el hígado puede excretar el fármaco a la bilis
- los procesos que pueden contribuir a una disminución
en la biodisponibilidad son el efecto de
primer paso o la eliminación
- Magnitud del efecto de primer paso hepático. Razón
de extracción (ER)
- ER = CL hepático / Q ; donde Q es
el flujo snaguíneo hepático
(en promediot 90 L/hora
- La biodisponibilidad sistémica (F) puede ser determinada
a partir del grado de absorción (f)
y la razón de extracción (ER):
Velocidad de absorción:
- la velocidad de absorción depende, en general, del
sitio de administración y de la forma farmacéutica.
- velocidad de absorción
de orden cero: independiente de la cantidad
de fármaco remanente en el intestino
- velocidad de absorción de primer orden: proporcional
a la cantidad de fármaco disuelto en el
tracto gastrointestinal
|
