- Introducción:
- Lipofilicidad: propiedad del fármaco que promueve
su paso a través de membranas biológicas
y facilita su llegada al sitio de acción.
Además disminuye su excreción..
- Nota: la excreción renal del fármaco "no modificado"
contribuye levemente a su eliminación: renal,
puesto que estos son generalmente
lipofílicos y por lo tanto fácilmente
reabsorbidos a través de las membranas
tubulares del riñón.
- La biotransformación de los fármacos a sustancias
más hidrofílicas, es necesaria para
su eliminación desde el organismo.
- Las reacciones de biotransformación producen sustancias
más polares, hidrofílicas y biológicamente
inactivas, que son más rápidamente
excretadas. Algunos metabolitos
pueden ser activados o retener la
actividad biológica del producto
natural o aún ser más tóxicos
que el agente agente primitivo.
- Los
mecanismos de biotransformación
de fármacos son descritos ya sea
como reacciones de Fase I y reacciones
de Fase II.
- Reacciones de Fase I y Reacciones de Fase II
-
- Características
de las reacciones de Fase I:
- El fármaco original es alterado introduciéndole
o exponiendolo a grupos funcionales
(-OH,-NH2, -SH)
- Los fármacos transformados por reacciones
de Fase I pueden perder
su actividad farmacológica.
- Los profármacos,inactivos,
son convertidos por reacciones
de Fase I a metabolitos biológicamente
activos.
- Los productos de
la Fase I pueden:
- ser directamente excretados por la orina.
- reaccionar
con compuestos endógenos para
formar conjugados solubles en
agua.
- Características
de la Fase II:
- El fármaco original o sus metabolitos pueden participar
en en reacciones de conjugación
que:
- forman un unión covalente entre un grupo funcional
del agente y:
- ácido glucurónico
- sulfato
- glutation
- amino ácidos
- acetato
- Los conjugados son:
- altamente polares
- generalmente inactivos
- una excepción a la regla es el metabolito de
glucurónico con morfina, que
es más potente que el compuesto
original.
- rápidamente excretados en la orina
- Los
conjugados de alto peso molecular:
- son excretados por la bilis
- la unión puede ser escindida por la
flora intestinal
- el fármaco original liberado puede regresar a
la circulación sistémica.
- este proceso se conoce como "circulación
enterohepática":
- retarda la eliminación del fármaco original
- prolonga el efecto del fármaco
Principales órganos comprometidos
en la biotransformación
- Principal Organo: Hígado
- Otros órganos metabolizantes:
- tracto gastrointestinal
- pulmones
- piel
- riñón
- Secuencia I
- Administración oral: (ej: isoproterenol,
meperidina, pentazocina )
- Absorbido intacto (a nivel del intestino delgado)
- Transportado hacia el hígado (sistema porta)
- Extensamente metabolizado (efecto de primer paso)
- Secuencia II
- Administración oral (clonazepam,
clorpromazina )
- Absorbido intacto (a nivel del intestino delgado)
- Extenso metabolismo intestinal ( que contribuye al
efecto total de primer paso)
Factores que
reducen la biodisponibilidad:
- Efecto de primer paso de los fármaco administrados
por vía oral
- La flora intestinal que puede metabolizar los fármacos
- Inestabilidad en el medio ácido gástrico
- Metabolización por enzimas digestivas
- Metabolización por enzimas de la pared intestinal
Sistema de oxidasas de función mixta
( sistema citocromo P 450 )
Reacciones
de Fase I
- Los microsomas han sido utilizados para estudiar las
oxidasas de función mixta
- Enzimas metabolizantes
de fármacos:
- Localizadas en las membranas del retículo endoplásmico
hepatico
- El retículo endoplásmico liso contiene las enzimas
responsables del metabolismo de
los fármacos
- La reacción:
- una molécula de oxígeno es concsumida por molécula
de sustrato.
- un átomo de oxígeno aparece en el producto y el
otro en la forma de agua.
- procesos de oxidación-reducción:
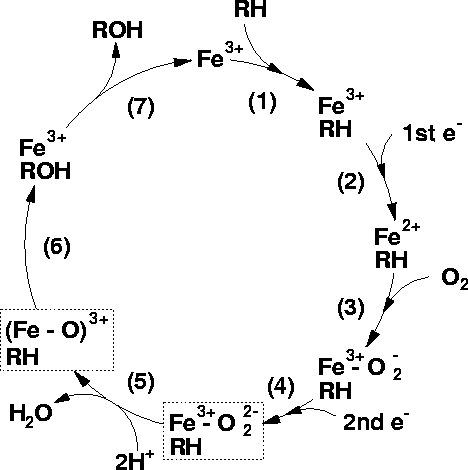
Ciclo del citocromo P450
La unión del sustrato al P450 provoca una reducción
en el potencial redox lo que favorece
la transferencia de un electrón desde
el NADH o NAPH.
- Primera reducción: El próximo paso en el ciclo es la
reducción del Fe3+ por
un electrón transferido desde el NADH
(o NAPH), a través de una cadena transportadora
de electrones.
- Unión del oxígeno una molécula de O2 se
une al Fe2+
formando
Fe2+-O2
- Segunda reducción: una segunda reducción
es requerida para la reacción
estequiométrica. Se ha determinado que
este es el paso de velocidad limitante
de la reacción
- Escisión de O2 : El
O2 reacciona con dos protones rompiéndose
la uión O-O formándose agua y el complejo Fe-O3+ .
- Formación de producto: El átomo de oxígeno ligado al
Fe es transferido al sustrato obteniéndose
la forma hidroxilada del sustrato.
- Liberación del producto: el producto es liberado del
sitio activo de la enzima la cual regresa
a su estado inicial.
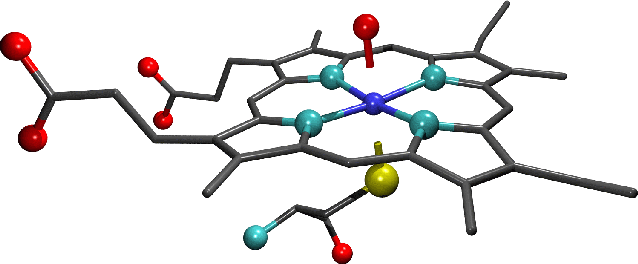
-
"Sitio activo del citocromo P450 . Note
la molécula de agua ( que es vista como
un único átomo de oxígeno) que forma
los seis ligandos axiales del hem. Los
átomos de oxígeno están en rojo, nitrógeno
en celeste, azufre en amarillo y Fe
en azul oscuro. Los átomos de carbono
se muestran sólo como uniones plomas
y sólo los hidrógenos han sido omitidos
por razones de claridad de la figura.
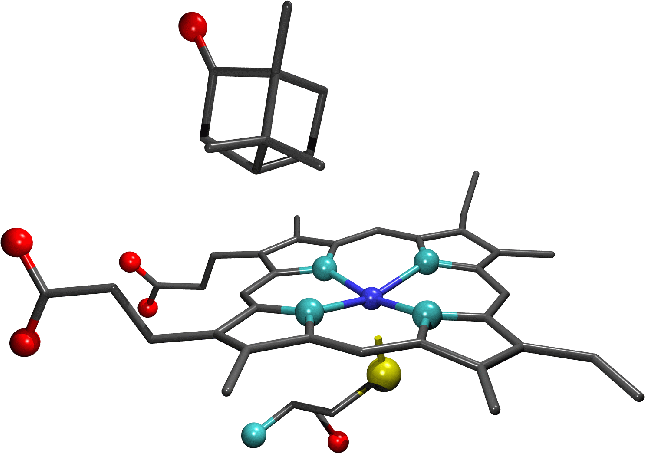
"Sitio activo mostrando la unión del alcanfor al
citocromo P450. Notese la ausencia
de moléculas de agua la cual forma el
ligando de seis axiales en la enzima
sin sustrato."
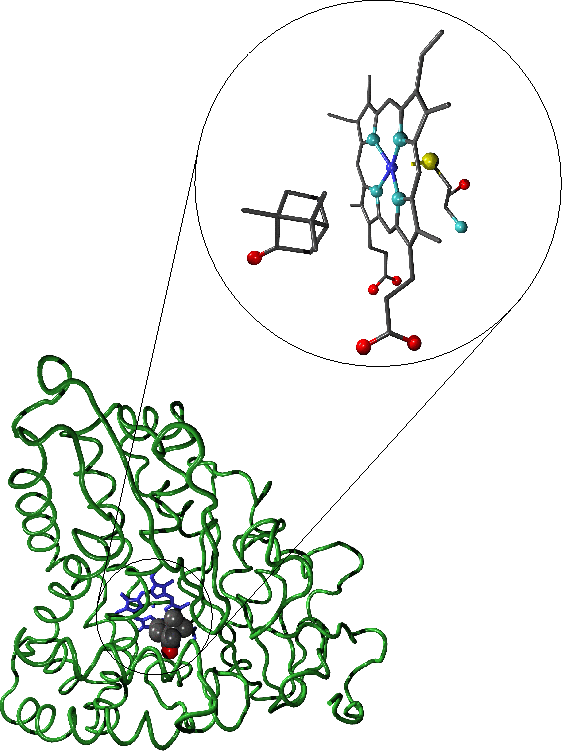
Representación de la unión del alcanfor. La región
aumentada del sitio activo muestra el
sustrato (alcanfor) el grupo hem y el
residuo de cisteína que forma el ligando
distal del hem. En esta representción,
el "esqueleto" de la proteína
es mostrado en verde el grupo
hem en azul y el sustrato es coloreado
de acuerdo al tipo de átomo. Los
átomosde´oxígeno se muestran en rojo,
carbono en gris, nitrógeno en celeste,
azufre en amarillo e hierro en azul.
- Inducción
enzimática del citocromo P450:
- Después de una administración repetida, algunos fármacos
inducen el citocromo P450 (aumenta la
cantidad de enzima). Esto es generalmente
debido a:
- Aumento en la velocidad de síntesis de la
enzima
- Disminución en la velocidad de degradación de la
enzima
- Inhibición
enzimática del citocromo P450:
- Ciertos fármacos inhiben competitivamente el
metabolismo al unirse al citocromo.
- . Ejemplos:
- Cimetidina ) (Bloqueador de los receptores
H2 usado en tratamiento
de la úlcera) y el ketoconazol (usado
en tratamiento de infección por
hongosl) se unen al hem del citocromo P450, reduciendo
el metabolismo de:
- testosterona
- otros fármacos co-administrados
- Mecanismo de Acción: inhbición competitiva
- Inactivación catalítica del citocromo P450.
- Los antibióticos del tipo macrólidos (eritromicina,
troleandomicina) son metabolizados
por el citocromo P450.
- los metabolitos
forman un complejo con el hem-Fe
del citocromo, produciendo un complejo
que es catalíticamente inactivo.
- El cloranfenicol, al ser metabolizado
por el citocromo P450, genera un
metabolito quelante que inactiva
el citocromo P450
- Otros inactivadores: Mecanismo de acción:
a nivel del grupo hem:
- etinil estradiol
- noretindrona
- espironolactona
- otros:
- propiltiouracilo
- etoclorvinol
Metabolismo
Fase II
Algunas reacciones de Fase II
|
Tipo de Conjugación |
Reactante endógeno |
Transferasa (Ubicación) |
Tipos de sustratos |
Ejemplos |
|
Glucuronisación |
Acido UDP glucurónico |
UDP glucuronosil transferasa
(microsomal) |
Fenoles alcoholes ácidos carboxílicos hidroxilaminas, sulfonamidas |
morfina, acetaminofeno, diazepam,
digitoxina, digoxina, meprobamato |
|
Acetilación |
Acetil-CoA |
N-Acetyl transferasa
(citosol) |
Aminas |
sulfonamidas, isoniazida, clonazepam,
dapsona, mescalina |
|
Conjugación con Glutatión |
glutatión |
GSH-S-transferasa (citosol,
microsomas) |
epóxidos, grupos nitro hidroxilaminas |
ácido etacrínico, bromobenzeno |
|
Conjugación con sulfato |
fosfoadnenosill
fosfosulfato |
Sulfotransferasa
(citosol) |
fenoles, alcoholes, aminas aromáticas |
estrona, 3-hidroxi cumarina,
acetaminofeno, metildopa |
|
Metilación |
S-Adenosyl-metionina |
transmetilasas (citosol) |
catecolaminas, fenoles, aminas,
histamina |
dopamina, adrenalina histamina,
tiouracilo, piridina |
- Reacciones Fase
II: Enzimas no microsomales
- Reaccones tipo:
- conjugación
- hidrolisis
- oxidación
- reducción
- Localización (enzimas no microsomales): principalmente
hepática ;también plasma y tracto gastrointestinal
- Enzimas no microsomales catalizan todas las
reacciones de conjugación excepto glucuronidaciones.
- Reacciones de conjugación:
- Conjugados:
- más polares
- fácilmente excretados
- generalmente inactivos
Conjugación:
- Compromete intermediarios de "alta energía"
y enzimas específicas de transferencia
(transferasas citosólicas o microsomales)
- La conjugación con glucurónico requiere de
las enzimas citocromo P450.
- ácido glucurónico: disponible desde la glucosa.
Este tipo de conjugación es:
- farmacológicamente inactivo
- más soluble en agua y más facilmente excretado
por la orina y bilis
- Transferasas:
- catalizan el acoplamiento de una sustancia endógena
con un fármaco
- derivado uridin-5'-difosfate (UDP) de ácido
glucurónico con un fármaco
- catalizan fármacos inactivados con un sustrato
endógeno
- por ejemplo: el derivado S-CoA del ácido benzoico
con un fármaco con un sustrato
endógeno.
- Toxicidad:
- Ciertas reacciones de conjugación pueden originar
productos tóxicos (hepatotóxicos)
- ejemplos:
- la acil glucuronidación de los antiinflamatorios
no esteroidales.
- La N-acetilation de la isoniazida
- Fármacos metabolizados a productos tóxicos:
- Hepatotoxicidad por acetaminofeno:
es un fármaco
seguro a dosis teraspéuticas
- A dosis terapéuticas:
- conjugados del tipo glucuronidación + conjugación
con sulfatos (95% de los metabolitos
excretados); 5% debido a una
vía de l citocromo P450 que
depende de conjugación
con glutatión (GSH)
- A altas dosis:
- Las vías de glucuronidación
y conjugación con sulfato se saturan.
- La vía dependiente del
citocrmo P450 es ahora la más importante
- con la depleción del glutatión hepático se
forman metabolitos electrofílicos,
reactivos y hepatotóxicos.
- Antidotos: N-acetilcisteína, cisteamina
- La N-acetilcssteíne: proteje a los pacientes
de la hepatotoxicidad
fulminante y muerte
por sobredosis de acetominofeno.
|