|
Algunos factores que influyen en la absorción de fármacos y su
biodisponibilidad.
Absorción
|
Principios de la absorción:
- Paso a través de membranas:
- Difusión pasiva (medio acuoso o lipídico): es
el proceso más
común
- Transporte activo: importante para algunos
fármacos,especialmente moleculas
grandes.
- I. Difusión acuosa
- en grandes componentes acuosos (e.g.,espacio
instersticial, citosol)
- a través de membranas epiteliales fuertemente
unidas
- a través del endotelio de los vasos sanguíneos
- a través de poros acuosos:permite la difusión
de moléculas con pesos moleculares
hasta 20.000-30.000
- Debido
a: gradiente de concentración
de fármacos
(descrito por Ley de Fick's).
- Existe una "fuerza" que obliga a
las moléculas a moverse
desde una alta concentración
hacia una menor concentración
según un movimiento aleatorio
de estas moléculas.
- Recuerda que las moléculas se mueven aleatoriamente,
de modo tal que una molécula
que se encontraba en el
lugar de baja concentración
puede moverse hacia el lugar
de alta concentración. Sin
embargo, en el balance,
es más probable que las
moléculas se muevan del lugar
de alta al de baja concentración.
Ley de Fick
- Esta ley describe el movimiento pasivo
de las moléculas
a favor de un
gradiente de
concentración
- Flujo (J) (moléculas por unidad
de
tiempo) =
(C1 -
C2) · (Area ·Coeficiente dePermeabilidad) /
Espesor |
- donde C1 es la mayor
concentrción y C2 es la menor concentración
- área = área a través de la cual ocurre
la difusión
- coeficiente de permeabilidad de movilización
del fármaco
en la vía de
difusión
- para difusión por lípidos: coeficiente
de partición lipido/agua c
- mayor
coeficiente,
mayor mobilidad
del fármaco
- este coefiente refleja cuan fácil el
fármaco
entra
a la
fase
lipídica
desde
la fase
acuosa.
- espesor: longitud de la vía de difusión
|
- El fármaco unido a proteínas plasmáticas
no puede penetrar los poros.
- Fármacos cargados pueden ser influídos
por campos de potencial eléctrico.{los
potenciales de membrana son importantes
en el transporte renal transtubular}
II. Difusión lipídica
- Es la más importante debido a:
- muchas barreras lipídicas separan los compartimentos
corporales
- El coeficiente lípido/agua describe
la facilidad con que el fármaco
se mueve entre el medio acuoso
y lipídico
- El
grado de ionización es un factor
importante pues los fármacos
cargados difunden con dificultad
a través del medio lipídico.
- El pH y el pKa del fármaco
son importantes para determinar
el grado de ionización,
el cual influye significativamente
el transporte .Razones de
las
formas solubles en lípidos
y en agua, son descritas,
tanto para ácidos como para
bases por la ecuación
de
Henderson-Hasselbalch .
- Forna sin carga:
liposoluble
- Forna cargada: soluble en
agua,
relativamente insoluble en lípidos (no atraviesa fácilmente
las membranas)
|
Ecuación de Henderson-Hasselbalch
Forma General: log
(protonada)/(no-protonada) = pKa - pH
- Para Acidos: pKa = pH
+ log ([HA] )/
[A-]
- note que si [A-] = [HA] entonces pKa
= pH + log (1) o (dado que log(1) = 0), pKa = pH
|
- Para Bases: pKa = pH + log (
[BH+])/ [B]
- note que si [B] = [BH+] entonces
pKa = pH + log (1) o (dado
que log(1) = 0), pKa = pH
|
1.-
Mientras menor el valor del pH en
relación
al valor
del
pKa , mayor será la fracción de fármaco protonado. Recuerde
que
un ácido
la fracción
protonado
no tiene
carga; mientras
que para
una base, la
forma protonada
tiene
carga.
2.-
Como resultado de ello un ácido
débil, en
medio ácido,
será más
liposoluble
debido a
que las
moléculas
no-cargadas
se mueven
más rápidamente
a través
de los lípidos
de las membranas.
3.-
Similarmente,
una base débil,
en pH alcalino,
será más liposoluble,
ya que a este
pH un protón
se disociará
de la molécula
dejándola sin
carga y ahora
con una mayor
capacidad de
atravesar membranas
lipídicas.
La difusión lipídica depende
de una adecuada
solubilidad
en lípido
La ionización de un fármaco
reduce su
capacidad
de atravesar
la membrana
lipídica
Muchos fármacos son ácidos
débiles
o bases
débiles
- Un ácido débil es una
molécula
neutra
que
se
disocia
en
un
anión
(cargado
negativamente)
y
un
protón
(ión
hidrógeno).Ejemplo:
- C8H7O2COOH
< >
C8H7O2COO-
+ H+
- aspirina neutra
(C8H7O2COOH) en
equilibrio
con
aspirina anión
(C8H7O2COO-
) y un protón (H+ )
- ácido débil
forma
protonada
neutra, más
liposoluble
- base débil:
una
molécula
neutra
que puede formar un catión (positivamente cargada)
conbinándose con un protón. Ejemplo:
- C12H11CIN3NH3+
< >
C12H11CIN3NH2
+ H+
- catión pirimetamina
(C12H11CIN3NH3+)
en equilibrio con pirimetamina neutra
(C12H11CIN3NH2)
y un protón (H+ )
- base débil:
forma protonatada --cargada, menos liposoluble
|
| Acidos débiles |
pKa |
Bases débiles |
pKa |
|
|
7.1 |
|
8.5 |
|
|
8.1 |
|
9.6 |
|
|
9.5 |
|
4.6 |
|
|
3.5 |
|
7.9 |
|
III. Transportadores especiales
Transportadores:
- El transporte activo describe un
proceso que requiere
energía que es saturable, que
probablemente es contra
un gradiente de concentración
y que comprende un número
finito de transportadores, por
lo tanto el proceso
se puede saturar cuando
todos las moléculas
de tranportadortes
están ocupadas.
- Difusión facilitada, aunque no
requiere energía es
también saturables (número
limitado de transportadores)
- Saturable (a diferencia de la difusión
pasiva) debido al número
limitado de sitios transportadores.
Una vez que estos sitios
están ocupados la velocidad
de transporte no puede
aumentar.
- Una
propiedad de los sistemas
transportadores es
que el proceso puede ser
inhibido por otras moléculas.
- Endocitosis:entrada a las células de sustancias
muy grandes (e.g., fierro vitamin B12
cada uno formando
complejos al unirse con proteínas y
pasan
a través de la pared
intestinal a la sangre)
- Los
neurotrasmisores son ejemplos
para la exocitocis:
- Después de la activación neuronal
eléctrica de los terminales
nerviosos dos pasos
pueden ser iniciados:
- Las vesículas de almacenamiento
que contienen el
neurotrasmisor,
se unen con la membrana
celular seguida
por:
- liberación o difusión del
contenido hacia
la región extracelular.
Resumen:
|
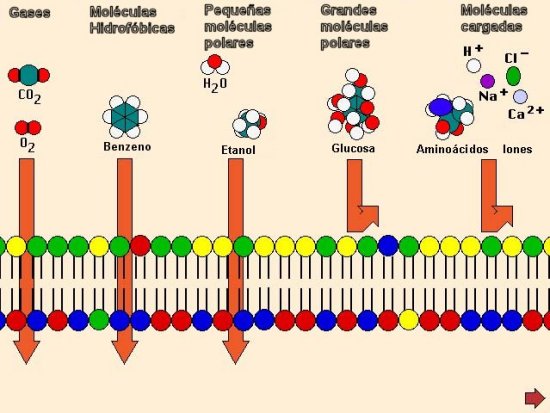
|

Grado
de absorción
- Una absorción incompleta, después
de la administración oral, es
bastante común:
Por ejemplo, sólo el 70% de una
dosis de digoxina alcanza
la circulación.
Factores:
- Una baja absorción gastrointestinal (GI)
- metabolismo por la flora gastrointestinal
- Las sustancias muy hidrofílicas, no
son bien absorbidas pues no
pueden atravesar el componente
lipídico de la membrana.
- Los fármacos excesivamente liposolubles
(hidrofóbicos) pueden no ser
lo suficientemente solubles para
atravesar la capa de agua cercana
a la membrana celular.
Atrapamiento
iónico
- Casi todos los fármacos son filtrados
a nivel del glomérulo:
- La mayoría de los fármacos, en
el estado liposoluble,
pueden ser reabsorbidos
por difusión pasiva.
- Para aumentar la excreción a este
nivel, se puede modificar
el pH urinario, a fin
de favorecer la forma
cargada del fármaco,
ya que estas no
son rápidamente absorbidas:
- Acidos
débiles: excretados más
rápidamente en pH alcalino
(se favorece la forma aniónica)
- Bases
débiles: excretadas más
rápidas en pH ácido (se
favorece la forma catiónica)
|
Vías de administración
|
Administración Oral
Más conveniente, más económica.
Desventajas:
- emesis (por irritación por causa
del fármaco de la mucosa
gastrointestinal)
- enzimas digestivas y/o acidez gástrica
puede destruir el fármaco
- absorción poco probable o inconsistente
debido a los alimento u
otros efectos del fármaco.
- metabolización del fármaco por la
flora intestinal
- Estado de ionización del fármaco:
- formas no-ionizada (liposoluble) favorecen
la absorción
- los ácidos débiles pueden estar
altamente ionizados
en el pH alcalino
intestinal (no se
favorece la absorción) pero
este efecto es contrarrestado por
la elevada superficie
de absorción
- los fármacos ácidos son mejor absorbidos
a nivel gástrico
- Efecto de primer paso
- Los fármacos absorbidos desde el
tracto gastrointestinal
llegan al hígado a través
de la vena porta, y luego
alcanzan la circulación
sistémica.
- Una alta metabolización y/o extracción
hepática puede dar
como resultado, para algunos
fármacos, que sólo alcancen
una pequeńa concentración
plasmática.
- Los fármacos con un pronunciado efecto
de primer paso, muestran
grandes diferencias en su
efecto farmacológico cuando
se compara la administración
oral con la endovenosa.
Administración transdérmica
Ventajas:
- mantenida, niveles plasmáticos terapéuticos
(oscilaciones plasmáticas asociadas con la admnistración intermitente)
- Evita las dificultades de una infusión
contínua.
- Poca incidencia de efectos laterales
(a pequeńas dosis)
- Buena cooperación y aceptación del paciente
Factores que contribuyen a una buena
absorción transdérmica.
- peso molecular < 1000
- rango de pH 5-9 en medio acuoso
- no hay liberación de histamina
- requerimientos diarios del fármaco <10
mg
Ejemplo de fármacos disponible para
administración transdérmica:
- escopolamina:-eventualmente puede
ocurrir tolerancia con una
pérdida de la acción terapéutica
- fentanil
- clonidina
- nitroglicerina:
eventualmente puede
ocurrir tolerancia con una
pérdida de la acción terapéutica
Administración Rectal
- Adminstración rectal proximal:
Absorción por las venas hemorroidales superiores que llegan
a la porta y luego al hígado (posible
efecto de primer paso) y finalmente
a la circulación sistémica.
- Administración rectal baja: el fármaco
puede alcanzar la circulación
sistémica sin pasar por el hígado.
- Generalmente origina una respuesta
farmacológica impredecible debido
a las razones anteriores.
- Probable
irritación de la mucosa rectal
Administración Parenteral
- Asegura una absorción efectiva del
fármaco
- La administración subcutánea e intramuscular
son más rápidas y predecibles
que la vía oral.
- Unica vía de administración apropiada
para:
- pacientes no cooperadores
- pacientes inconcientes
- Factores
que determinan la velocidad
de absorción sistémica.:
- área de las membranas capilares absorbentes
- solubilidad del fármaco en el líquido
intersticial
- canales acuosos (endotelio vascular
) favorecen la velocidad de difusión de los fármacos, independiente
de su solubilidad lipídica
- Ventajas de la administración endovenosa:
- Se
obtienen rápidos y precisos
niveles plasmáticos ( no hay
efecto de primer paso)
- Los fármacos irritantes se administran
más comodamente (los vasos
sanguíneos son relativamente
insensibles); fármaco rápidamente
diluído (particularmente si es administrado en grandes
venas de los brazos)
|
EFECTO DE
PRIMER PASO
Eliminación de primer paso:
- Secuencia de transporte:
- paso a través de la pared intestinal hacia
la circulación portal
- la sangre de la porta lleva el fármaco
al hígado
- el fármaco puede luego alcanzar la circulación
sistémica
- la biodisponibilidad puede ser afectada por
estos pasos
- la metabolización de fármacos puede ocurrir en
la pared intestinal o en
la sangre
- la metabolización de fármacos puede ocurrir en
el hígado
- el hígado puede excretar fármacos hacia la bilis
- los procesos que contribuyen a la disminución
de la biodisponibilidad
es la pérdida por primer
paso o la eliminación.
- Magnitud
del efecto de primer paso
hepático. Razón de extracción
(ER):
- ER = CL hepático/ Q ; donde Q es el flujo sanguíneo
hepático (aproximadamente 90 L /hora {1500 ml/min})
- La biodisponibilidad sistémica del fármaco
(F) puede ser determinada
a partir del grado
de absorción (f) y la
razón de extracción (ER):
Razón de Extracción, Vías de Administración y Efecto de
Primer Paso.
- Algunos fármacos que presentan una alta razón
de extracción son administrados
por vía oral.
- Algunos ejemplos: desipramina, imipramina, meperidina, propranolol, amitriptilina , isoniazida.
- Algunos fármacos con baja biodisponibilidad no
son administrados oralmente
debido a una toxicidad metabólica.
La lidocaína es un ejemplo (toxicidad
a nivel del SNC,
convulsiones)
- Fármacos con elevada razón de extracción hepática
muestran variaciones de
biodisponibilidad entre
diferentes pacientes debido
a diferencias en:
- función hepática
- flujo sanguíneo
- presencia de enfermedad hepática
Fármacos con
baja extracción hepática
- fenitoína
- diazepam
- digitoxina
- clorpropamida
|
- teofilina
- tolbutamida
- warfarina
|
- Se evita efecto de primer paso:
- vía sublingual (e.g. nitroglicerina). Acceso
directo a la circulación
sistémica
- vía transdérmica
- por uso de supositorios en el recto inferior {si
el supositorio es depositado
o se mueve hacia el
interior, la absorción
puede ocurrir a través
de las venas hemorroidales
superiores, que lo llevan
al hígado}
- por inhalación puede: puede ocurrir un
efecto de primer paso
pulmonar por excreción
o por metabolización.
|
|
